Genie De Novo Protein Design by Equivariantly Diffusing Oriented Residue Clouds
这篇文章的方法简单有效
之前的方法要构建C-alpha的旋转矩阵的话,需要构建SE(3)或者SU(2)的扩散模型,十分复杂。然后这篇文章通过利用Frenet-Serret frames,就可以根据C-alpha的坐标生成对应的旋转矩阵,也就是说只需要找到C-alpha即可,那么又回到了传统的ddpm模型。
Intro
Generative modeling trilemma
- quality
- mode coverage
- sampling time
其实intro没讲啥,就是引入了一种新的建模方式(cloud,其实就是旋转矩阵+平移向量)achieve sota
Method
FS frames构建过程如下,可以看到只需要输入序列C-alpha的坐标就好

模型
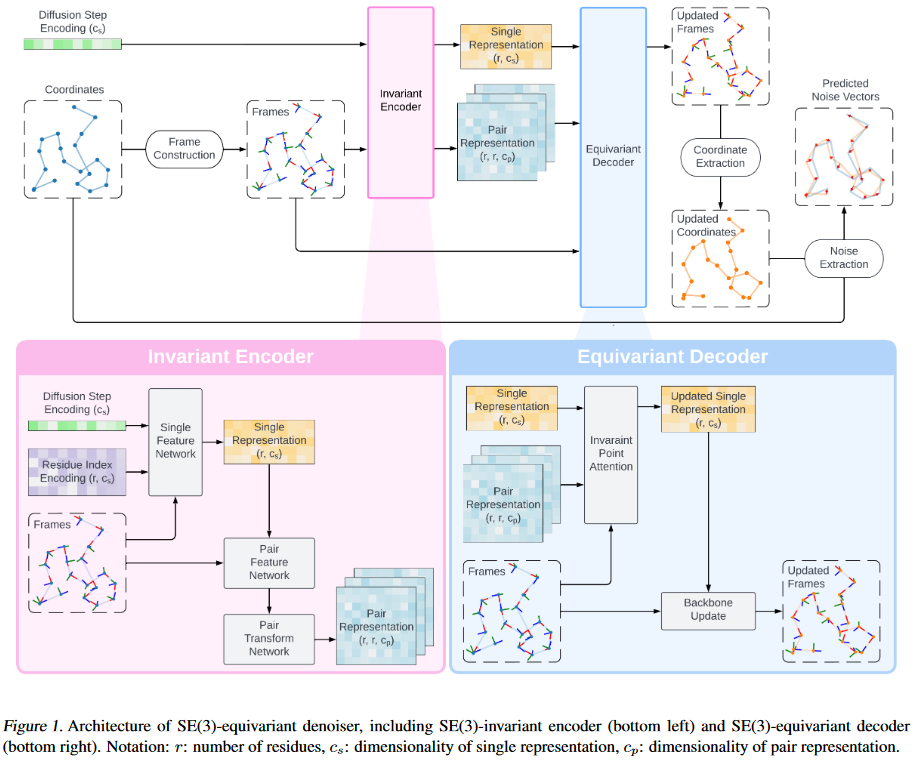
看上去就是AF2的structure module(不过这篇文章画这么大的模型图真的合适吗?又不是重点)
训练
其实就是ddpm的目标函数,不过code中用的是RMSD计算距离

Results
Comparison with short models (50-128)
Designability
方法:Genie采样得到的结构->8条ProteinMPNN生成序列(temperature=0.1)-> OmegaFold(或者ESMFold)预测结构,和采样的结构算TM-score
| Metric | Genie | ProtDiff | FoldingDiff |
|---|---|---|---|
| scTM>0.5 | 81.5% | 5.1% (11.8%) | 19.6% (22.7%) |
| scTM>0.5&pLDDT>70 | 58.3% | 3.2% | 17.7% |
Diversity
- 看secondary structure elements(用P-SEA识别二级结构)
对所有scTM>0.5&pLDDT>70的蛋白质,画出其二级结构的分布,发现Genie分布最diverse
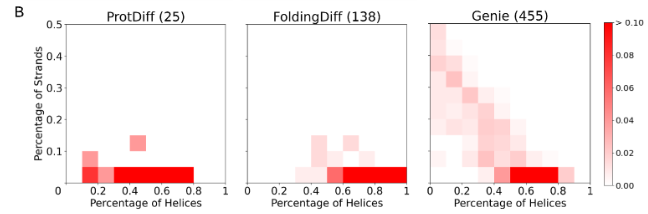
- 看一个蛋白质和confident domain上的蛋白质的最高TM-score(越小说明越diverse)
| Metric | Genie | ProtDiff | FoldingDiff |
|---|---|---|---|
| maximum TM-score | 0.561 ± 0.086 | 0.583 ± 0.115 | 0.668 ± 0.178 |
Novelty
算和training set中的maximum TM-score
| Metric | Genie | ProtDiff | FoldingDiff |
|---|---|---|---|
| maximum TM-score<0.5 | 21.5% | 4% | 20.3% |
(这里看上去其实Genie生成的很大一部分蛋白质都是跟training set中一致的,怀疑过拟合了,也只比FoldingDiff好一点)
Comparison with short models (50-256)
和RFdiffusion,FrameDiff比
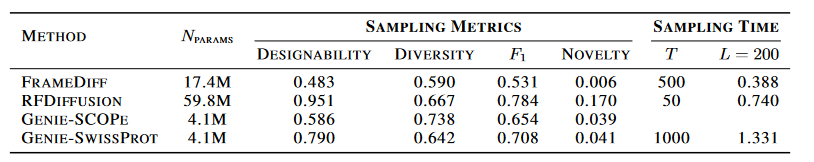
Designability
ProteinMPNN+ESMFold: scRMSD<2 and pLDDT<70
Diversity
看下二级结构分布。(看上去FrameDiff和RFDiffsion好像更好)
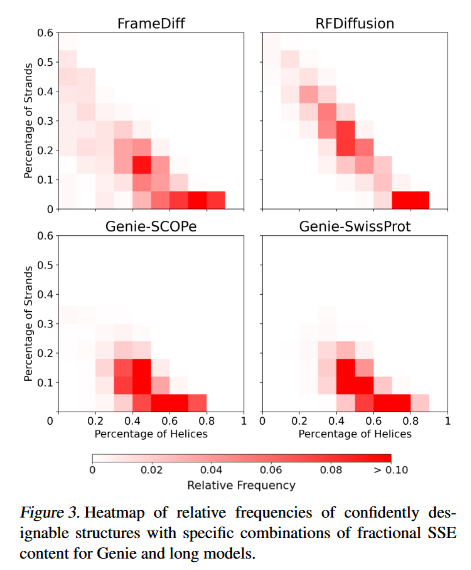
并且考虑三级结构的diversity,给所有的可设计蛋白质结构聚合(根据TMscore>0.6)

Novelty
对比PDB中的蛋白质,如果scRMSD<2 and pLDDT>70 and Maximum TM <0.5则认为是novel
结论
其实是被RFdiffusion吊打的,但是可以说RFdiffusion是pretrain过的,但是Genie是从头开始训练的